PeptideAtlas Adds Millions of Microproteins to Human Proteome
Scientists have expanded the human proteome by identifying millions of novel microproteins and peptideins using advanced mass spectrometry. The PeptideAtlas database now includes these previously undiscovered protein fragments, offering new avenues for biological research.
Researchers have significantly expanded the known human proteome by identifying millions of novel microproteins and peptideins, thanks to advanced mass spectrometry techniques and the comprehensive PeptideAtlas database. This discovery, detailed in a recent study, effectively adds a vast new layer to our understanding of human biology and disease.
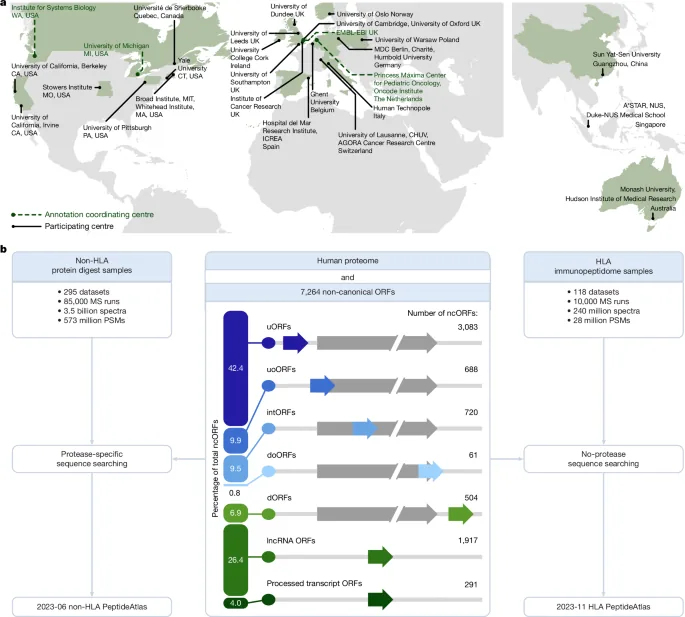
The updated Human PeptideAtlas database, version 2023-06, now incorporates data from 295 ProteomeXchange datasets, encompassing 1.17 billion MS/MS spectra. These spectra were analyzed using the MSFragger software, with search parameters tailored to each specific dataset. The analysis focused on a 2023-02 THISP level 4 database, which includes over 7,000 open reading frames (ORFs) identified through ribosome profiling, alongside other potentially translatable sequences. Generic artifactual modifications like methionine oxidation and N-terminal acetylation were considered, along with semi-tryptic settings to maximize peptide identification.
Statistical validation for each experiment was rigorously conducted using the Trans-Proteomic Pipeline (TPP), including tools like PeptideProphet and iProphet. Results were then mapped to the human proteome and genome using ProteoMapper and the ENSEMBL toolkit. Supplementary Table 15 provides detailed false discovery rate (FDR) metrics at various levels, including PSM, peptide, and protein, for the core proteome and newly identified sequences, many of which are putative ncORFs (non-coding open reading frames).
Unveiling the 'Dark Proteome'
A parallel effort focused on the Human HLA PeptideAtlas, adding 240 million MS/MS spectra from 9,776 runs. This build, using a no-enzyme search mode, identified numerous peptides that do not conform to traditional tryptic fragmentation patterns. To estimate the accuracy of ncORF detections, researchers employed a target-decoy entrapment approach. This method involves creating scrambled decoy protein sequences at a 1:1 ratio with target sequences, allowing the system to estimate false positive identifications. While not perfect, this technique is widely accepted as a standard in the field for ensuring the reliability of novel peptide identifications.
The integration of these millions of microproteins and peptideins significantly broadens the scope of the human proteome, often referred to as the 'dark proteome' due to the vast number of proteins previously undetected or uncharacterized. These smaller protein fragments, sometimes overlooked in traditional proteomic studies, can play crucial roles in cellular signaling, regulation, and disease pathogenesis. Understanding their functions is vital for developing new diagnostic and therapeutic strategies.
Proteins identified in the PeptideAtlas are categorized based on the strength of evidence. Canonical proteins require at least two unique, non-nested peptides of a minimum length, covering a substantial amino acid sequence. Non-core canonical proteins meet these criteria but map outside the standard core proteome. The database also includes categories for marginally distinguished, weakly supported, and indistinguishable proteins, reflecting varying levels of evidence. For integration with Human Proteome Project (HPP) metrics, only entries linked to the ~20,000 neXtProt or UniProtKB/Swiss-Prot protein-coding genes can achieve canonical status.
Despite extensive efforts to minimize errors, the database acknowledges the presence of some false positives, particularly among detections mapping to proteins unlikely to be observed. To address this, a manual inspection process was implemented for peptides corresponding to ncORFs. These were categorized as 'excellent,' 'good,' 'false positive,' 'close but false positive,' or 'low information,' providing a qualitative assessment of the identification's reliability. The best peptide-spectrum match for each is available via Universal Spectrum Identifiers (USI) for online viewing, ensuring transparency and enabling further validation by the scientific community.